 |
|
Research
The primary interest of the Huang lab is to study the molecular basis of genetic syndromes, to apply the discoveries from rare diseases to common conditions and to develop treatments for genetic diseases. Currently, we are focusing on the following areas.
1. The role of TBX3 in breast cancer and human emryonic stem (hES) cells: TBX3 is a T-box transcription factor. Mutations of TBX3 cause Ulnar-Mammary syndrome characterized by hypoplasia or absence of the mammary glands. Our lab is one of the first groups to show that overexpression of TBX3 plays an important role in breast cancer. Our study shows that TBX3 is overrexpressed in primary breast cancer tissues. Mechanistically, we find that TBX3 interacts with HDACs to inhibit downstream target gene expression, such as p14ARF. In addition, we find that TBX3 regulates a large group of genes in breast cancer. Our current research aims to optimize the clinical relevance of this data working in parallel with animal and breast cancer tissues. Recently, we have also found that TBX3 plays a very important role in hES cell differentiation. This finding may further our understanding of TBX3 function. 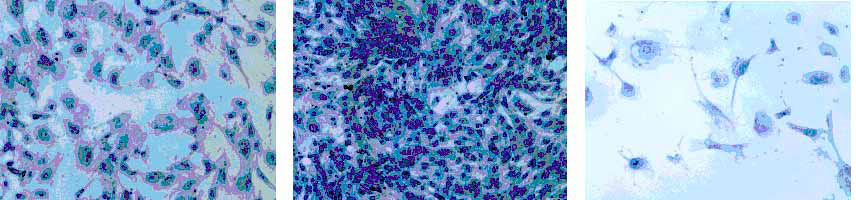
Roles of TBX3 and TBX3+2a on Senescence of Mouse Embryo Fibroblast Cells
|
|
|
2. The Genetic basis of optic atrophy and inductible pluripotent stem cell (iPSC) therapy: We have worked with many families affected by auosomal dominant inherited optic atrophy. In collaboration with Dr. Arnold Star in the Department of Neurology, we find that the OPA1 gene mutation H445R causes loss of vision and hearing. Using electrophysiological analysis, we find that this mutation causes asynchronous cochlear conduction, suggesting a novel mechanism of optic atrophy. To study the function of OPA1 and the molecular mechanismsof optic atrophy, we created a drosophila model of POA1. We found that the dOpa1 somatic mutation caused an increase in reactive oxygen species (ROS) production and mitochondrial framentation. Our group shows that antioxidants can partially reverse the glossy eye phenotype, further suggesting that ROS plays an important role in cell death. Together, these results show that dOpa1 mutations cause cell loss by two distinct pathogenic pathways. This study provides novel insights into the pathogenesis of optic atrophy and demonstrates the promise of antioxidants as therapeutic agents for this condition. Recently, our lab is actively engaged in iPS cell therapy. We have succesffully differentiated iPS cells into retinal ganglion cells. |
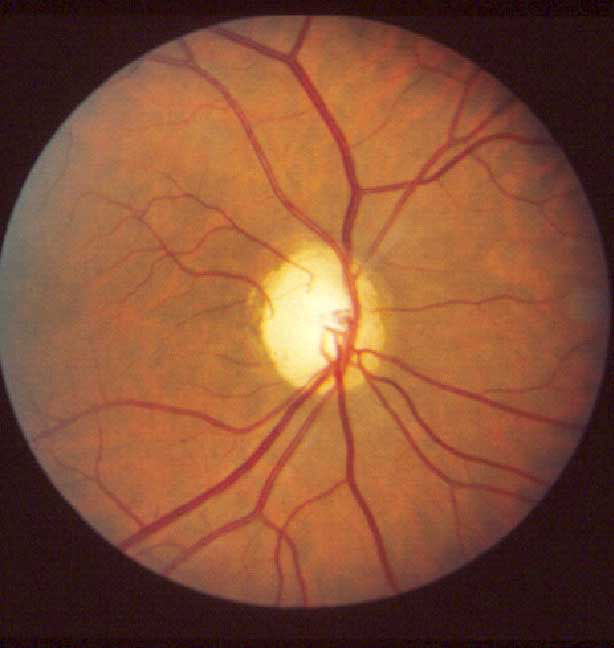 Optic Atrophy |
| 3. Identifying the disease-causing gene associated with noncompaction of the ventricular myocardium (spongy heart): We are currently studying a family with balanced translocation with this condition and are also performing a linkage study for a large pedigree with this disease. Currently, our lab is using next-generation sequencing technology to identify the new disease-causing gene. |
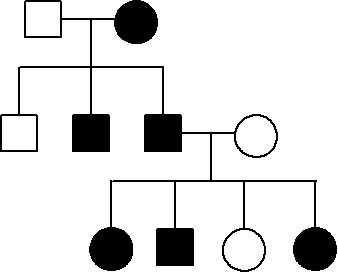
Autosomal Dominant INVM
|
|
4. Identification of the disease-causing gene for Lenz Microphthalmia Syndrome (LMS) using whole genome exome sequencing technology: LMS is a rare condition characterized by small eyes/no eye and multiple congenital anomalies such as small brain and mental deficiency, abnormal ear, teeth, digits, skeletal and/or genitourinary tract. In this study, we have used a very powerful technology, next generation sequencing, to search for the disease-causing gene in patients with LMS. Identification of disease-causing genes associated with LMS has significantly facilitated our understanding of this condition and can translate into clinical applications. Since LMS affects multiple organ systems, understanding the gene associated with LMS may open a window for the investigation of other common conditions and human development.
|
|
| 5. The intracellular pathway to study TBX5: TBX5 is a T-box transcription factor. Mutations of TBX5 cause Holt-Oram syndrome characterized by congenital heart diseases and limb anomalies. By studying the intracellular network of TBX5, including the upstream transcription factor that controls TBX5 expression and the cofactors that interact with TBX5 and its downstream targets, we anticipate identifying many genes associated with congenital heart disease, the most common congenital malformation in humans, which contribute significantly to mobility and mortality in pediatric populations. | 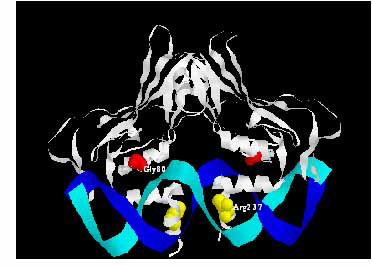
Mutations of TBX5 cause Holt-Oram Syndrome
|